
Integrated Computational Materials Engineering (ICME)
LAMMPS Intrinsic Stacking-Fault Energy
Questions / Comments
Email: Mark A. Tschopp, mark.tschopp@gatech.edu
Abstract
This tutorial demonstrates how to create an intrinsic stacking-fault in FCC metals, taking Ni as an example, and how to estimate its energy (SFE) using molecular dynamics (MD). The stacking fault is created using two different methods. In the first, the shearing of the crystal on {111} slip plane along the <112> direction, as in the actual deformation process in metals. The second method involves the stacking fault is created following a thought process whereby a layer one atom thick parallel to {111} plane is removed and the material put back together. LAMMPS is used to perform MD calculations while OVITO, visualization software for atomic structures, is used to visualize the stacking fault using the centro-symmetry and energy per atom parameters generated by LAMMPS.
Author(s): Richard Glaze IV, Firas Akasheh*, Mark A. Tschopp
(*) Mechanical Engineering Department, Tuskegee University, Tuskegee, AL 36088
Introduction
Generally, metals have crystalline structures having long-range order consisting of a repetitive unit cell. Nonetheless, crystalline materials are never perfect and normally have defects. There are three types of defects in metals: point, line and planar. This tutorial focuses on a planar defect called intrinsic stacking fault commonly observed in face-centered cubic (FCC) metals. A stacking fault is an irregularity in the planar stacking sequence of atoms in a crystal. The natural stacking arrangement for FCC metals on the closed packed {111} plane is ABCABCAB…, but if an intrinsic stacking fault is introduced, the stacking arrangement changes to ABCBCABC…. It is as if one closed plane, A in this case, has been removed, disrupting the otherwise perfect stacking. Such departure from perfect order increases the energy of the material. The stacking-fault energy (SFE) quantifies this additional energy and is measured in terms of energy per unit area of the stacking fault. The SFE has a significant effect on the deformation behavior of metals. Metals with low stacking fault energy tend to have more twinning, more extended (partial) dislocations which have less mobility and less propensity to cross slip. This in turn affects hardening, ductility, climb, and texture development during deformation. Additionally, stacking faults provide a strong barrier to dislocation motion which becomes more significant in nanoscale metallic structures.
Description of Simulation
A simulation box consisting of pure Ni is generated by LAMMPS. The x, y, and z axes of the simulation box are aligned with the [1 1 2], [-1 1 0], and [-1 -1 1] crystal directions, respectively. As such the {111} plane lies in the x-y plane which will facilitate the process of generating the stacking fault. The box size is 4.3 x 4.3 x 3.7 nm in the x, y, and z directions, respectively, and contains 3600 atoms. The model has periodic boundary in the x and y directions, while free boundary conditions are applied in the z direction. Two input script files were written to simulate the creation of an intrinsic stacking fault using two different methods. In the first method, the top half of the crystal is displaced as a rigid body with respect to the bottom half in the negative x direction by a calculated value. The calculated value is the distance from 1 “dip” (low energy stable position) in between atoms to the next nearest one, which is along the Burgers’ vector of the partial dislocation. The second method follows the thought process where a layer of atoms is simply removed and the material allowed to be joined back together and the structure relaxed. After creating the stacking fault, the input file (script) calculates the stacking-fault energy using the following equation:
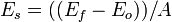
where,
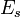 = stacking-fault energy
= stacking-fault energy
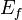 = the potential energy of the system after creating the stacking-fault
= the potential energy of the system after creating the stacking-fault
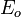 = the potential energy of the system before creating the stacking-fault
= the potential energy of the system before creating the stacking-fault
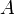 = area of the stacking-fault
= area of the stacking-fault
Input
LAMMPS input script for FCC
As mentioned above, two LAMMPS script files were created, “stackint1.txt” and “stackint2.txt”. To run the script, open a command window (using CMD command in the case of Windows systems) and change the directory to the location of your LAMMPS program. At the prompt type the command "lmp_win_no-mpi.exe < filename.txt" in a Windows environment and hit return. Here, "lmp_win_no-mpi.exe" refers to the LAMMPS executable and filename is any of stackint1 or stackint2. Below is shown the script of these two file:
stackint1
# Input file for Stack Fault Energy surface of Nickel
# Richard Glaze, 2014
# --------------------- INITIALIZAITION ---------------------
clear
units metal
dimension 3
boundary p p s
atom_style atomic
variable latparam1 equal 3.52
variable x_displace equal -1*(${latparam1}/sqrt(6))
variable xdim equal ${latparam1}*sqrt(6)/2*10
variable ydim equal ${latparam1}*sqrt(2)/2*10
# --------------------- ATOM DEFINITION ---------------------
lattice fcc ${latparam1}
region 1 block -.001 ${xdim} -.001 ${ydim} -.001 16.5 units box
region 2 block -.001 ${xdim} -.001 ${ydim} 16.5 36.5808 units box
region whole block 0 ${xdim} 0 ${ydim} 0 36.5808 units box
#the -.001 in the -x and -y and lower limit of region 1 are only to overcome a simple numerical issue but can be considered 0
create_box 2 whole
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
create_atoms 1 region 1
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
create_atoms 2 region 2
# --------------------- FORCE FIELDS ---------------------
pair_style eam/alloy
pair_coeff * * FeCuNi.eam.alloy Ni Ni
# --------------------- SETTINGS ---------------------
compute peratom all pe/atom
compute eatoms all reduce sum c_peratom
compute csym all centro/atom fcc
thermo 1
thermo_style custom step pe c_eatoms
dump 1 all custom 1 dump.relax.1.* id type xs ys zs c_peratom c_csym
run 0
#this command creates a model of the script before the displacement and minimization occur
variable E equal "c_eatoms"
variable Eo equal $E
#variable E equal "c_eatoms" computes the initial energy of the model before any sliding is done
#E is necessary to store the initial energy in Eo
group bot region 1
group top region 2
displace_atoms top move ${x_displace} 0.0 0.0 units box
#displace_atoms is the command neccessary to move atoms into the next stacking arrangement (ie: A -> B)
fix 1 all setforce 0 0 NULL
min_style cg
minimize 1e-10 1e-10 1000 1000
variable Ef equal "c_eatoms"
variable Cf equal 1.60217657e-16
variable A equal (${xdim}*${ydim})*1e-20
variable SFE equal ((${Ef}-${Eo})*${Cf})/${A}
#variable Ef equal "c_eatoms" computes the final energy of the system after sliding is done
#variable A is the area of the Stacking fault plane
#variable Cf is the conversion factor of electro volts to millijoules
#variable SFE is the stacking-fault energy of the system
####################################
# SIMULATION DONE
print "All done"
print "Initial energy of atoms = ${Eo} eV"
print "Final energy of atoms = ${Ef} eV"
print "Stacking-fault energy = ${SFE} mJ/m^2"
stackint2
# Input file for Stack Fault Energy surface of Nickel
# Richard Glaze, 2014
# --------------------- INITIALIZAITION ---------------------
units metal
dimension 3
boundary p p s
atom_style atomic
variable latparam1 equal 3.52
variable x_displace equal -1*(${latparam1}/sqrt(6))
variable z_displace equal -1*(${latparam1}/sqrt(3))
variable xdim equal ${latparam1}*sqrt(6)/2*10
variable ydim equal ${latparam1}*sqrt(2)/2*10
variable Ecoh equal -4.45026
#Ecoh is the cohesive energy of Nickel
# --------------------- ATOM DEFINITION ---------------------
lattice fcc ${latparam1}
region 1 block -.001 ${xdim} -.001 ${ydim} -.001 15.4839 units box
region 2 block -.001 ${xdim} -.001 ${ydim} 15.4839 17.5161 units box
region 3 block -.001 ${xdim} -.001 ${ydim} 17.5161 36.5808 units box
region whole block 0 ${xdim} 0 ${ydim} 0 36.5808 units box
#the -.001 in the -x, -y, and lower limit of region 1 are only to overcome a simple numerical issue but can be considered 0
create_box 3 whole
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
create_atoms 1 region 1
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
create_atoms 2 region 2
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
create_atoms 3 region 3
# --------------------- FORCE FIELDS ---------------------
pair_style eam/alloy
pair_coeff * * FeCuNi.eam.alloy Ni Ni Ni
# --------------------- SETTINGS ---------------------
compute peratom all pe/atom
compute eatoms all reduce sum c_peratom
compute csym all centro/atom fcc
thermo 1
thermo_style custom step pe c_eatoms
dump 1 all custom 1 dump.relax.1.* id type xs ys zs c_peratom c_csym
run 0
variable N equal count(all)
variable No equal $N
#variable N equal count(all), counts the total number of atoms in the system
#the total number of atoms is stored to the variable No
#This command creates a model of the script before the displacement and minimization occur
variable E equal "c_eatoms"
variable Eo equal $E
#variable E equal "c_eatoms" computes the initial energy of the model before any sliding is done
#E is necessary to store the initial energy in Eo
group bot region 1
group middle region 2
group top region 3
delete_atoms group middle
#The delete_atoms command is used to remove the designated layer of atoms
variable Nf equal $N
#variable N is now set to the number of atoms after the removal of a layer which is assigned to Nf
displace_atoms top move 0.0 0.0 ${z_displace} units box
#displace_atoms is used here to merge to the two remaining regions
fix 1 all setforce 0 0 NULL
min_style cg
minimize 1e-10 1e-10 1000 1000
variable Ef equal "c_eatoms"
variable A equal (${xdim}*${ydim})*1e-20
variable Cf equal 1.60217657e-16
variable Er equal (${No}-${Nf})*${Ecoh}
variable SFE equal ((${Ef}-(${Eo}-${Er}))*${Cf})/${A}
#variable Ef equal "c_eatoms" computes the final energy of the system after removal is done
#variable A is the area of the Stacking fault plane
#variable Cf is the conversion factor of electro volts to millijoules
#variable Er is the total energy of the removed atoms
#variable SFE is the stacking-fault energy of the system
####################################
# SIMULATION DONE
print "All done"
print "Initial energy of atoms = ${Eo} eV"
print "Final energy of atoms = ${Ef} eV"
print "The Stacking-fault energy = ${SFE} mJ/m^2"
Output for method 1 using stackint1
LAMMPS logfile
The log file for first script, stackint1.txt should now look like this:
LAMMPS (1 Jul 2012)
# Input file for Stack Fault Energy surface of Nickel
# Richard Glaze, 2014
# --------------------- INITIALIZAITION ---------------------
units metal
dimension 3
boundary p p s
atom_style atomic
variable latparam1 equal 3.52
variable x_displace equal -1*(${latparam1}/sqrt(6))
variable x_displace equal -1*(3.52/sqrt(6))
variable xdim equal ${latparam1}*sqrt(6)/2*10
variable xdim equal 3.52*sqrt(6)/2*10
variable ydim equal ${latparam1}*sqrt(2)/2*10
variable ydim equal 3.52*sqrt(2)/2*10
# --------------------- ATOM DEFINITION ---------------------
lattice fcc ${latparam1}
lattice fcc 3.52
Lattice spacing in x,y,z = 3.52 3.52 3.52
region 1 block -.001 ${xdim} -.001 ${ydim} -.001 16.5 units box
region 1 block -.001 43.111019472983934 -.001 ${ydim} -.001 16.5 units box
region 1 block -.001 43.111019472983934 -.001 24.890158697766473 -.001 16.5 units box
region 2 block -.001 ${xdim} -.001 ${ydim} 16.5 36.5808 units box
region 2 block -.001 43.111019472983934 -.001 ${ydim} 16.5 36.5808 units box
region 2 block -.001 43.111019472983934 -.001 24.890158697766473 16.5 36.5808 units box
region whole block 0 ${xdim} 0 ${ydim} 0 36.5808 units box
region whole block 0 43.111019472983934 0 ${ydim} 0 36.5808 units box
region whole block 0 43.111019472983934 0 24.890158697766473 0 36.5808 units box
#the -.001 in the -x and -y and lower limit of region 1 are only to overcome a simple numerical issue but can be considered 0
create_box 2 whole
Created orthogonal box = (0 0 0) to (43.111 24.8902 36.5808)
1 by 1 by 1 MPI processor grid
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
lattice fcc 3.52 orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
Lattice spacing in x,y,z = 5.74814 4.97803 6.09682
create_atoms 1 region 1
Created 1800 atoms
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
lattice fcc 3.52 orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
Lattice spacing in x,y,z = 5.74814 4.97803 6.09682
create_atoms 2 region 2
Created 1800 atoms
# --------------------- FORCE FIELDS ---------------------
pair_style eam/alloy
pair_coeff * * FeCuNi.eam.alloy Ni Ni
# --------------------- SETTINGS ---------------------
compute peratom all pe/atom
compute eatoms all reduce sum c_peratom
compute csym all centro/atom fcc
thermo 1
thermo_style custom step pe c_eatoms
dump 1 all custom 1 dump.relax.1.* id type xs ys zs c_peratom c_csym
run 0
WARNING: No fixes defined, atoms won't move (verlet.cpp:54)
Memory usage per processor = 5.65496 Mbytes
Step PotEng eatoms
0 -15803.062 -15803.062
Loop time of -2.15368e-008 on 1 procs for 0 steps with 3600 atoms
Pair time (%) = 0 (-0)
Neigh time (%) = 0 (-0)
Comm time (%) = 0 (-0)
Outpt time (%) = 0 (-0)
Other time (%) = -2.15368e-008 (100)
Nlocal: 3600 ave 3600 max 3600 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 4422 ave 4422 max 4422 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 292200 ave 292200 max 292200 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 584400 ave 584400 max 584400 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 584400
Ave neighs/atom = 162.333
Neighbor list builds = 0
Dangerous builds = 0
#this command creates a model of the script before the displacement and minimization occur
variable E equal "c_eatoms"
variable Eo equal $E
variable Eo equal -15803.061682140291
#variable E equal "c_eatoms" computes the initial energy of the model before any sliding is done
#E is necessary to store the initial energy in Eo
group bot region 1
1800 atoms in group bot
group top region 2
1800 atoms in group top
displace_atoms top move ${x_displace} 0.0 0.0 units box
displace_atoms top move -1.437033982432798 0.0 0.0 units box
#displace_atoms is the command neccessary to move atoms into the next stacking arrangement (ie: A -> B)
fix 1 all setforce 0 0 NULL
min_style cg
minimize 1e-10 1e-10 1000 1000
WARNING: Resetting reneighboring criteria during minimization (min.cpp:173)
Memory usage per processor = 6.3416 Mbytes
Step PotEng eatoms
0 -15798.914 -15798.914
1 -15799.279 -15799.279
2 -15799.398 -15799.398
3 -15799.441 -15799.441
4 -15799.492 -15799.492
5 -15799.524 -15799.524
6 -15799.536 -15799.536
7 -15799.539 -15799.539
8 -15799.543 -15799.543
9 -15799.547 -15799.547
10 -15799.558 -15799.558
11 -15799.574 -15799.574
12 -15799.58 -15799.58
13 -15799.583 -15799.583
14 -15799.585 -15799.585
15 -15799.588 -15799.588
16 -15799.589 -15799.589
17 -15799.591 -15799.591
18 -15799.591 -15799.591
19 -15799.591 -15799.591
20 -15799.593 -15799.593
21 -15799.593 -15799.593
22 -15799.595 -15799.595
23 -15799.595 -15799.595
24 -15799.596 -15799.596
25 -15799.596 -15799.596
26 -15799.596 -15799.596
27 -15799.596 -15799.596
28 -15799.596 -15799.596
29 -15799.596 -15799.596
30 -15799.596 -15799.596
31 -15799.596 -15799.596
32 -15799.596 -15799.596
Loop time of 4.87928 on 1 procs for 32 steps with 3600 atoms
Minimization stats:
Stopping criterion = energy tolerance
Energy initial, next-to-last, final =
-15798.9141278 -15799.596273 -15799.5962737
Force two-norm initial, final = 3.25725 0.00227164
Force max component initial, final = 0.110875 8.68516e-005
Final line search alpha, max atom move = 0.0625 5.42822e-006
Iterations, force evaluations = 32 134
Pair time (%) = 2.77416 (56.8559)
Neigh time (%) = 0 (0)
Comm time (%) = 0.0100004 (0.204956)
Outpt time (%) = 2.00712 (41.1355)
Other time (%) = 0.0880036 (1.80362)
Nlocal: 3600 ave 3600 max 3600 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 4422 ave 4422 max 4422 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 288400 ave 288400 max 288400 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 576800 ave 576800 max 576800 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 576800
Ave neighs/atom = 160.222
Neighbor list builds = 0
Dangerous builds = 0
variable Ef equal "c_eatoms"
variable Cf equal 1.60217657e-16
variable A equal (${xdim}*${ydim})*1e-20
variable A equal (43.111019472983934*${ydim})*1e-20
variable A equal (43.111019472983934*24.890158697766473)*1e-20
variable Sfe equal ((${Ef}-${Eo})*${Cf})/${A}
variable Sfe equal ((-15799.596273718327-${Eo})*${Cf})/${A}
variable Sfe equal ((-15799.596273718327--15803.061682140291)*${Cf})/${A}
variable Sfe equal ((-15799.596273718327--15803.061682140291)*1.6021765700000001e-016)/${A}
variable Sfe equal ((-15799.596273718327--15803.061682140291)*1.6021765700000001e-016)/1.0730401163050707e-017
#variable Ef equal "c_eatoms" computes the final energy of the system after sliding is done
#The final energy is stored to the variable Ef
#variable Sfe is the stacking-fault energy of the system
###################################
# SIMULATION DONE
print "All done"
All done
print "Initial energy of atoms = ${Eo} eV"
Initial energy of atoms = -15803.061682140291 eV
print "Final energy of atoms = ${Ef} eV"
Final energy of atoms = -15799.596273718327 eV
print "Stacking-fault energy = ${Sfe} mJ/m^2"Stacking-fault energy = 51.742671078034931 mJ/m^2
Output for method 2 using stackint2
LAMMPS logfile
The log file for second script, stackint2.txt should now look like this:
LAMMPS (1 Jul 2012)
# Input file for Stack Fault Energy surface of Nickel
# Richard Glaze, 2014
# --------------------- INITIALIZAITION ---------------------
units metal
dimension 3
boundary p p s
atom_style atomic
variable latparam1 equal 3.52
variable x_displace equal -1*(${latparam1}/sqrt(6))
variable x_displace equal -1*(3.52/sqrt(6))
variable z_displace equal -1*(${latparam1}/sqrt(3))
variable z_displace equal -1*(3.52/sqrt(3))
variable xdim equal ${latparam1}*sqrt(6)/2*10
variable xdim equal 3.52*sqrt(6)/2*10
variable ydim equal ${latparam1}*sqrt(2)/2*10
variable ydim equal 3.52*sqrt(2)/2*10
variable Ecoh equal -4.45026
#Ecoh is the cohesive energy of Nickel
# --------------------- ATOM DEFINITION ---------------------
lattice fcc ${latparam1}
lattice fcc 3.52
Lattice spacing in x,y,z = 3.52 3.52 3.52
region 1 block -.001 ${xdim} -.001 ${ydim} -.001 15.4839 units box
region 1 block -.001 43.111019472983934 -.001 ${ydim} -.001 15.4839 units box
region 1 block -.001 43.111019472983934 -.001 24.890158697766473 -.001 15.4839 units box
region 2 block -.001 ${xdim} -.001 ${ydim} 15.4839 17.5161 units box
region 2 block -.001 43.111019472983934 -.001 ${ydim} 15.4839 17.5161 units box
region 2 block -.001 43.111019472983934 -.001 24.890158697766473 15.4839 17.5161 units box
region 3 block -.001 ${xdim} -.001 ${ydim} 17.5161 36.5808 units box
region 3 block -.001 43.111019472983934 -.001 ${ydim} 17.5161 36.5808 units box
region 3 block -.001 43.111019472983934 -.001 24.890158697766473 17.5161 36.5808 units box
region whole block 0 ${xdim} 0 ${ydim} 0 36.5808 units box
region whole block 0 43.111019472983934 0 ${ydim} 0 36.5808 units box
region whole block 0 43.111019472983934 0 24.890158697766473 0 36.5808 units box
#the -.001 in the -x, -y, and lower limit of region 1 are only to overcome a simple numerical issue but can be considered 0
create_box 3 whole
Created orthogonal box = (0 0 0) to (43.111 24.8902 36.5808)
1 by 1 by 1 MPI processor grid
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
lattice fcc 3.52 orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
Lattice spacing in x,y,z = 5.74814 4.97803 6.09682
create_atoms 1 region 1
Created 1600 atoms
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
lattice fcc 3.52 orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
Lattice spacing in x,y,z = 5.74814 4.97803 6.09682
create_atoms 2 region 2
Created 200 atoms
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
lattice fcc 3.52 orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
Lattice spacing in x,y,z = 5.74814 4.97803 6.09682
create_atoms 3 region 3
Created 1800 atoms
# --------------------- FORCE FIELDS ---------------------
pair_style eam/alloy
pair_coeff * * FeCuNi.eam.alloy Ni Ni Ni
# --------------------- SETTINGS ---------------------
compute peratom all pe/atom
compute eatoms all reduce sum c_peratom
compute csym all centro/atom fcc
thermo 1
thermo_style custom step pe c_eatoms
dump 1 all custom 1 dump.relax.1.* id type xs ys zs c_peratom c_csym
run 0
WARNING: No fixes defined, atoms won't move (verlet.cpp:54)
Memory usage per processor = 5.65496 Mbytes
Step PotEng eatoms
0 -15803.062 -15803.062
Loop time of -4.86616e-008 on 1 procs for 0 steps with 3600 atoms
Pair time (%) = 0 (-0)
Neigh time (%) = 0 (-0)
Comm time (%) = 0 (-0)
Outpt time (%) = 0 (-0)
Other time (%) = -4.86616e-008 (100)
Nlocal: 3600 ave 3600 max 3600 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 4422 ave 4422 max 4422 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 292200 ave 292200 max 292200 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 584400 ave 584400 max 584400 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 584400
Ave neighs/atom = 162.333
Neighbor list builds = 0
Dangerous builds = 0
variable N equal count(all)
variable No equal $N
variable No equal 3600
#variable N equal count(all), counts the total number of atoms in the system
#the total number of atoms is stored to the variable No
#This command creates a model of the script before the displacement and minimization occur
variable E equal "c_eatoms"
variable Eo equal $E
variable Eo equal -15803.061682140291
#variable E equal "c_eatoms" computes the initial energy of the model before any sliding is done
#E is necessary to store the initial energy in Eo
group bot region 1
1600 atoms in group bot
group middle region 2
200 atoms in group middle
group top region 3
1800 atoms in group top
delete_atoms group middle
Deleted 200 atoms, new total = 3400
#The delete_atoms command is used to remove the designated layer of atoms
variable Nf equal $N
variable Nf equal 3400
#variable N is now set to the number of atoms after the removal of a layer which is assigned to Nf
displace_atoms top move 0.0 0.0 ${z_displace} units box
displace_atoms top move 0.0 0.0 -2.032272947547483 units box
#displace_atoms is used here to merge to the two remaining regions
fix 1 all setforce 0 0 NULL
min_style cg
minimize 1e-10 1e-10 1000 1000
WARNING: Resetting reneighboring criteria during minimization (min.cpp:173)
Memory usage per processor = 6.3416 Mbytes
Step PotEng eatoms
0 -14908.862 -14908.862
1 -14909.227 -14909.227
2 -14909.345 -14909.345
3 -14909.396 -14909.396
4 -14909.461 -14909.461
5 -14909.475 -14909.475
6 -14909.482 -14909.482
7 -14909.488 -14909.488
8 -14909.5 -14909.5
9 -14909.507 -14909.507
10 -14909.517 -14909.517
11 -14909.525 -14909.525
12 -14909.53 -14909.53
13 -14909.536 -14909.536
14 -14909.539 -14909.539
15 -14909.54 -14909.54
16 -14909.54 -14909.54
17 -14909.541 -14909.541
18 -14909.542 -14909.542
19 -14909.542 -14909.542
20 -14909.543 -14909.543
21 -14909.543 -14909.543
22 -14909.543 -14909.543
23 -14909.543 -14909.543
24 -14909.543 -14909.543
25 -14909.544 -14909.544
26 -14909.544 -14909.544
27 -14909.544 -14909.544
28 -14909.544 -14909.544
29 -14909.544 -14909.544
30 -14909.544 -14909.544
31 -14909.544 -14909.544
32 -14909.544 -14909.544
33 -14909.544 -14909.544
34 -14909.544 -14909.544
35 -14909.544 -14909.544
36 -14909.544 -14909.544
37 -14909.544 -14909.544
38 -14909.544 -14909.544
39 -14909.544 -14909.544
40 -14909.544 -14909.544
41 -14909.544 -14909.544
42 -14909.544 -14909.544
43 -14909.544 -14909.544
44 -14909.544 -14909.544
Loop time of 6.13735 on 1 procs for 44 steps with 3400 atoms
Minimization stats:
Stopping criterion = energy tolerance
Energy initial, next-to-last, final =
-14908.8618058 -14909.543956 -14909.5439565
Force two-norm initial, final = 3.25725 0.0022227
Force max component initial, final = 0.110875 7.55982e-005
Final line search alpha, max atom move = 0.125 9.44978e-006
Iterations, force evaluations = 44 174
Pair time (%) = 3.37519 (54.9942)
Neigh time (%) = 0 (0)
Comm time (%) = 0.0170026 (0.277036)
Outpt time (%) = 2.66416 (43.409)
Other time (%) = 0.0809981 (1.31976)
Nlocal: 3400 ave 3400 max 3400 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 4176 ave 4176 max 4176 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 270800 ave 270800 max 270800 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 541600 ave 541600 max 541600 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 541600
Ave neighs/atom = 159.294
Neighbor list builds = 0
Dangerous builds = 0
variable Ef equal "c_eatoms"
variable A equal (${xdim}*${ydim})*1e-20
variable A equal (43.111019472983934*${ydim})*1e-20
variable A equal (43.111019472983934*24.890158697766473)*1e-20
variable Cf equal 1.60217657e-16
variable Er equal (${No}-${Nf})*${Ecoh}
variable Er equal (3600-${Nf})*${Ecoh}
variable Er equal (3600-3400)*${Ecoh}
variable Er equal (3600-3400)*-4.4502600000000001
variable SFE equal ((${Ef}-(${Eo}-${Er}))*${Cf})/${A}
variable SFE equal ((-14909.543956477508-(${Eo}-${Er}))*${Cf})/${A}
variable SFE equal ((-14909.543956477508-(-15803.061682140291-${Er}))*${Cf})/${A}
variable SFE equal ((-14909.543956477508-(-15803.061682140291--890.05200000000002))*${Cf})/${A}
variable SFE equal ((-14909.543956477508-(-15803.061682140291--890.05200000000002))*1.6021765700000001e-016)/${A}
variable SFE equal ((-14909.543956477508-(-15803.061682140291--890.05200000000002))*1.6021765700000001e-016)/1.0730401163050707e-017
#variable Ef equal "c_eatoms" computes the final energy of the system after removal is done
#variable A is the area of the Stacking fault plane
#variable Cf is the conversion factor of electro volts to millijoules
#variable Er is the total energy of the removed atoms
#variable SFE is the stacking-fault energy of the system
####################################
# SIMULATION DONE
print "All done"
All done
print "Initial energy of atoms = ${Eo} eV"
Initial energy of atoms = -15803.061682140291 eV
print "Final energy of atoms = ${Ef} eV"
Final energy of atoms = -14909.543956477508 eV
print "The Stacking-fault energy = ${SFE} mJ/m^2"The Stacking-fault energy = 51.747407860943149 mJ/m^2
Post-Processing
Visualization
The atomic structure during the simulation can be visualized using specialized software, in our case OVITO (www.ovito.org; developer: Alexander Stukowski). First, open Ovito. Then, select File, then Open Local Files. Then, navigate to the directory of the dump files LAMMPS created. The first dump file, dump.relax.1.0 should be the visualization of the model before sliding. An image of what you should see is located in Figure 1. Different colors are used to distinguish the two halves of the crystal which will be displaced with respect to each other. Now, if you would like the view the model after the sliding or the removal of a layer is done, go back to File and Open Local Files. Then, locate the last dump file created and double click it or single click it and press Open. The image that should appear can be seen in Figure 2. Notice how the stacking order is disrupted at the mid plane in comparison to the perfect stacking shown in Figure 1. To visually see the stacking fault you have two options. In both options, you use the Color coding option under the “Add modification…” drop box in Ovito. The first option is by Color coding by the property titled “c_peratom”. This option shows you the energy per atom. The other option is to color code the model by “c_csym”. This means they are color coded by their center of symmetry. For both of these options you are required to create a Slice which would get rid of the surface atoms as their center of symmetry and energy are too high. To do this you have to choose the “Slice” option under the “Add modification…” drop box. Set the normal to x and y to be 0 while the normal to the z direction is set to 1. Then, set the slice width high enough that it includes all but 6 layers. Finally, adjust the distance until it only excludes the 6 outer layers. After this step, you can resume to Color coding by “c_peratom” or “c_csym”. Make sure the Color coding option is above the Slice option in the area marked “Modifications”, then press “Adjust range”. Figure 3 shows the model color coded by the energy per atom. Figure 4 has the model color coded by the atoms’ center of symmetry. A close up of the normal stacking arrangement (B-C-A) can be found in Figure 5.
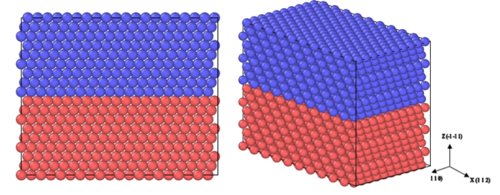
Figure 1: Model before slipping or deleting with 3600 atoms created by two layers of 1800 atoms.

Figure 2: Model after the sliding or deleting (depending on which script you chose to run) is complete.
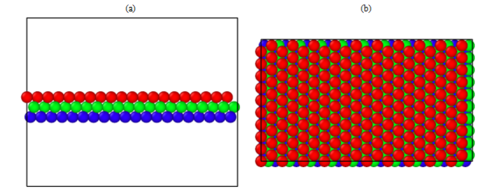
Figure 3: (a) side (b) top view of the perfect stacking arrangement A-C-B before slipping or deleting
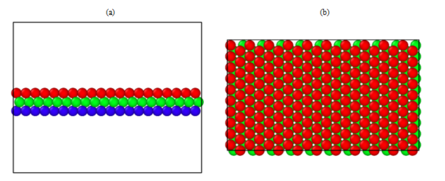
Figure 4: (a) side (b) top view of the stacking arrangement after sliding/deleting. Notice the gap in between atoms in the top view because the first and third layer are aligned proving it’s an B-C-B arrangement.
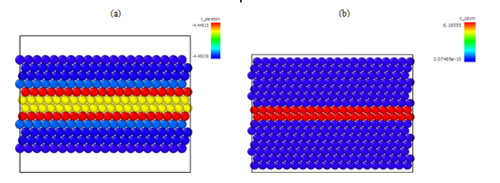
Figure. 5: Visualization of the intrinsic stacking fault, (a) color coded by the centrosymmetry parameter, and (b) color coded by energy per atom. The layers in the middle where the stacking fault was created have higher energies than surrounding layer
Acknowledgments
We would like to acknowledge the support to this work by the National Science Foundation, HBCUUP-RIA program, Program Manager Dr. Claudia Rankins, Award No. HRD-1137587. Additionally, the technical and logistical support of CAVS and HPC2 of Mississippi State University is gratefully acknowledged.